



用于T细胞恶性肿瘤的CAR-T细胞疗法
摘要:嵌合抗原受体T细胞(CAR-T)疗法彻底改变了B细胞淋巴样肿瘤的治疗,并在某些情况下改善了疾病结果。因此,已经引入了六个FDA批准的商业CAR-T细胞产品,这些产品针对在恶性B细胞或浆细胞上优先表达的抗原,用于B细胞淋巴瘤、B-ALL和多发性骨髓瘤的治疗。

这些治疗成功激发了CAR-T细胞疗法在其他血液肿瘤,包括T细胞恶性肿瘤中的应用。然而,CAR-T细胞疗法在T细胞肿瘤中的成功明显受限,这是由于存在一些限制因素,例如:1)正常T细胞和CAR-T细胞与恶性细胞之间共享的共同抗原,决定了兄弟相残事件和严重的T细胞再生障碍;2)用于CAR转导的CAR-T细胞被恶性T细胞污染。异体CAR-T产品可以避免肿瘤污染,但引发了与免疫不兼容性相关的其他问题。
尽管存在这些限制,在过去几年中,在针对T细胞恶性肿瘤的CD7和CD5靶向CAR-T细胞疗法方面取得了显著进展。
介绍
T细胞急性淋巴细胞性白血病(T-ALL)约占成人ALL的25%和儿童ALL的10-15%。T-ALL来源于T细胞发育过程中胸腺祖细胞的白血病转化,这是通过累积遗传异常实现的。
根据分化阶段和一些膜抗原的表达,T-ALL被分类为早期/前皮质、皮质和成熟。随着分子遗传学分析(下一代测序)和细胞遗传学研究的出现,T-ALL根据这些遗传异常的存在被分类。在T-ALL中观察到两种类型的遗传异常:A型异常,特征是依赖于染色体重排或缺失的转录因子的异位激活;B型改变,与需要额外的遗传病变以实现完全白血病转化有关。根据遗传异常的存在,已确定四种T-ALL亚型,包括早期胸腺细胞祖细胞(ETP)/不成熟ALL、TLX、TLX1/NKX2.1和TAL/LMO。ETP-ALL的特征是表达造血干细胞标记物如CD34、异常HOX4和MEF2C基因表达、IL7信号级联中的复发突变和高BCL2蛋白表达。TLX亚群的特征是表达γ/δ T细胞受体、激活TLX3或HOXA转录因子的驱动遗传事件。TLX1/NKX2.1亚群的特征是涉及TLX1或NKX2.1转录因子的驱动遗传事件和TLX重排病例中NUP214-ABL1融合的发生。TAL/LMO亚群的特征是表达成熟细胞膜标记物、TAL1和LMO2转录因子的驱动激活和PTEN的复发突变。
过去三十年来,包括T-ALL在内的ALL治疗取得了显著进展,总体生存率得到了持续改善,特别是对于15岁以下的儿童。然而,成人T-ALL患者的生存率明显低于儿童和青少年。此外,复发/难治性疾病患者的预后较差,生存率在10-25%之间。
T细胞淋巴瘤是一种涉及T细胞恶性转化的非霍奇金淋巴瘤;已确定四种主要亚型,包括外周T细胞淋巴瘤、皮肤T细胞淋巴瘤(Sezary综合征和蕈样霉菌病)、间变大细胞淋巴瘤和血管免疫母细胞性T细胞淋巴瘤。在理解这些疾病的分子发病机制方面取得了相当大的进展,从而改善了它们的治疗;然而,对于难治性或复发性疾病的患者,结果通常较差。
嵌合抗原受体(CAR)是一种新型的基于细胞的免疫疗法,显示出相当的疗效。CAR-T细胞被设计为专门识别细胞上表达的主要组织相容性复合体(MHC)无关的抗原,并因此杀死该细胞。CAR-T细胞的生产包括三个步骤:首先从患者或捐赠者那里获得健康细胞,然后使用基因组技术(如慢病毒基因转导和/或基因编辑)对T细胞进行工程改造,最后注入武装的CAR-T细胞以识别和杀死癌细胞。CAR-T细胞治疗在治疗B细胞恶性肿瘤方面取得了相当的成功,如B细胞淋巴瘤和B急性淋巴细胞性白血病。这些在治疗几种血液系统恶性肿瘤方面的发展激发了对T细胞恶性肿瘤的CAR-T细胞治疗的探索;然而,将CAR-T细胞治疗扩展到T细胞恶性肿瘤尤其具有挑战性,因为正常和恶性细胞之间共表达许多细胞膜抗原靶标。
基于CD7靶向的T-ALL CAR-T细胞治疗。CD7是T-ALL细胞高表达的膜抗原。CD7是一种分子量为40 KDa的细胞膜糖蛋白,属于免疫球蛋白超基因家族。CD7被认为是治疗T-ALL和T细胞淋巴瘤的良好潜在靶标。
最近,工程化的表达抗CD7的CAR-T淋巴细胞被用作T-ALL和T细胞淋巴瘤治疗的治疗剂。然而,由于CD7在正常T淋巴细胞和NK细胞上表达,T-淋巴细胞细胞膜上CD7的不受限制的表达将决定兄弟相残的杀戮;因此,生成靶向CD7的CAR-T细胞需要在它们被工程化以编码抗CD7的CAR之前,取消T-淋巴细胞膜上CD7的阻断。
为了开发靶向CD7的CAR-T细胞,已经使用了各种方法来抑制/阻断T细胞上CD7的表达:基因编辑、蛋白质阻断剂和自然选择。
基于基因编辑的CD7 CAR-T细胞。使用了两种基因编辑来生成CD7 CAR-T细胞:CRISPR/CAS9基因编辑和基础编辑基因编辑。
CRISPR/CAS9基因编辑。CRISPR/CAS9基因编辑系统涉及两个组成部分:caspase 9(CAS9)切割DNA,以及导向RNA(CRISPR,成簇的规律间隔短回文重复序列),将CAS9导向特定DNA序列的水平:因此,设计针对特定DNA序列的导向RNA,可以使用CAS9引入DNA的双链断裂,从而产生特定的基因敲除。临床前研究表明,CD7KO CD7 CAR-T细胞可以防止兄弟相残,增殖,并发挥针对恶性T白血病细胞的特定抗肿瘤活性。
可以使用自体T细胞或异体T细胞来生成CD7 CAR-T细胞。从患者那里获得的自体T细胞用于生成CD7 CAR-T细胞是有限的:从患者那里获得适当数量的健康细胞用于准备CAR-T细胞的困难;生成CAR-T细胞所需的程序持续时间;用于CAR-T细胞生成的T细胞可能被白血病或淋巴瘤T细胞污染的风险;自体、个性化CAR-T细胞制剂的相当大的成本。
Allo-CAR-T细胞没有这些限制;然而,这些CAR-T细胞有其他问题,主要与免疫兼容性有关:宿主免疫系统可能会拒绝被识别为非自身的异体CAR-T细胞;供体T细胞对宿主的移植物抗宿主病(GvHD)的发展。
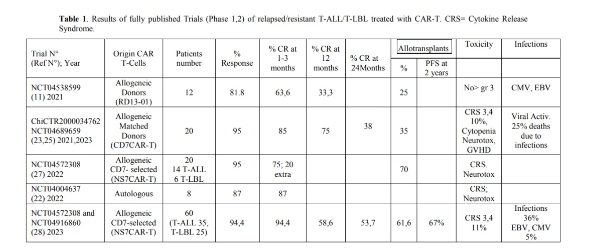
基因编辑可以用来敲除T细胞受体基因,以减少T细胞的免疫不兼容性,消除人类白细胞抗原(HLA)II类基因和CD52。使用这种方法,谢等人最近报告了“通用”CAR-T细胞CD7-/-、TRAC-/-、CD7UCAR.的开发。这些细胞在体外有效增殖,并特异性诱导原发性T-ALL细胞的杀伤,分泌大量促炎细胞因子;此外,这些CAR-T细胞也能显著减少肿瘤负荷并延长T-ALL模型小鼠的生存期。胡等人报告了首次使用CD7靶向CAR-T细胞(RD13-01)进行的临床试验,这些细胞经过基因改造(敲除CD7/TRAC/RFX5相关基因),以抵抗同种异体杀伤、GvHD和异体排斥,并增强抗肿瘤活性。在这项I期试验中,共有12名患者(11名T-ALL/T-淋巴瘤患者和1名表达CD7的AML患者)接受了这些CAR-T细胞治疗。11 输注后4周,9/11的患者显示出OR,7/11的患者达到CR;3名患者接受了异体HSCT。中位时间为10.5个月时,4名患者仍然处于CR状态。(表1)张等人最近报告了一项I期研究的结果,该研究涉及7名T-ALL和3名T-LBL患者接受RD13-01 CAR-T细胞治疗, 80%的患者实现了CR,7/8的应答患者MRD阴性;有趣的是,三名之前自体CD7 CAR-T细胞治疗失败的患者在接受CD7异体HSCT治疗后实现了CR,4/6的患者直到315天仍然无进展。七名患有髓外病变(EMD)的患者中有四名在中位第30天获得了EMD CR。2/6的患者复发且没有CD7丢失,随后死亡。只有一名患者经历了3级细胞因子释放综合征,一名患者经历了3级神经毒性。这些观察结果支持进一步研究,以更好地定义RD13-01产品在治疗T-ALL和T-LBL患者中的安全性和有效性。
Leedom及其同事报告了WU-CART-007的开发,这是一种用于T细胞恶性肿瘤的同种异体CAR-T细胞疗法,使用正常T细胞通过CRISPR/CAS9基因编辑敲除CD7和TRAC(通过GUIDE-Seq控制可能的非目标事件的发生),随后通过慢病毒载体转导CAR,表达高亲和力和高特异性的抗CD7,细胞扩增和耗尽残留的TCRA/B+细胞。WU-CART-007细胞通过CD7靶向在体外和体内都展现出强大的抗肿瘤活性。使用WU-CART-007细胞,Ghobadi等人报告了I/II期研究WU-CART-007-1001的结果,该研究涉及12名R/R T-ALL或T-LBL患者,接受四个剂量水平(每次输注100、300、600、900百万细胞)的治疗。WU-CART-007显示出可管理的毒性,25%的患者出现与治疗相关的不良事件;在剂量水平<2的患者中,CRR为43%。Li等人报告了基于慢病毒转化的allo-CAR-T细胞GC027的开发,该细胞表达抗CD7,正常健康T淋巴细胞中的TCRα和CD7基因被CRISPR/CAS9基因编辑系统沉默。最初在两名难治性/复发性T-ALL患者中探索了GC027 CAR-T细胞的安全性和有效性,两名患者均实现了完全反应。最近,同一作者报告了涉及12名R/R T-ALL患者的GC027 CAR-T细胞的扩展研究结果:11/12的患者在CAR-T细胞输注后一个月内迅速清除了白血病性T淋巴母细胞并达到CR(CR率91.7%)。四名EMD患者中有三名显示出病变的完全缓解。输注的GC027细胞在体内迅速扩增,并在输注后5-10天达到扩增高峰;在大多数患者中,GC0278细胞在输注后4周不可检测。值得注意的是,一名患者的PFS超过3年。毒性概况是可管理的。
基因编辑。基因编辑是一种新兴的基因组技术,它能够在特定的基因位点上以高特异性、精确度和效率产生可编程的单碱基对变化。在这项技术中,腺嘌呤碱基编辑器(EBEs)和胞嘧啶碱基编辑器(CBEs)结合一个单链DNA脱氨酶酶与核酸酶CAS9,分别在特定的基因组靶位点安装A·T到G·C或C·G到T·A的点突变。由于ABEs和CBEs都不引起双链断裂,它们产生高效的靶向编辑,显著降低了核酸酶编辑中观察到的复杂基因组重排的风险。
利用这项技术,DiOrio等人开发了7CAR8,这是一种通过引入四个同时的碱基编辑:CD7、TCRα、CD52和PD1,产生的针对CD7的异体CAR-T。在临床前研究中,7CAR8-T细胞在体外和体内T-ALL模型中显示出高效性。
2022年,CD7 CAR-T基础编辑临床试验(ISRCTN 15323014)开始。在这个试验中,健康捐赠者的T细胞在TCRβC1和TCRβC2、CD7和CD52水平上进行了基础编辑,然后转导了编码识别CD7的CAR的慢病毒载体;使用这个程序,产生了基础编辑的CAR7(BE-CAR7)T细胞库。这项研究的I期将涉及10名患有难治性/复发性T-ALL或其他T细胞恶性肿瘤的儿童;这些患者首先接受了淋巴细胞耗竭,随后接受了0.2x10^6到2x10^6 CE-CAR7 T细胞每公斤体重的输注;在28天分子缓解的患者接受了异体HSCT,随后使用HSCT前使用的条件方案耗尽任何持续存在的BER-CAR7细胞。最近报告了前3名接受治疗的患者的结果:第一名患者,一名13岁的女孩,在异体HSCT后复发T-ALL,在接受BE-CAR7输注后实现了分子缓解,然后从她原来的捐赠者那里接受了移植,成功实现了免疫重建和持续的白血病缓解;第二名患者出现了致命的真菌并发症;第三名患者在BE-CAR7细胞输注后实现了分子缓解,随后在缓解期接受了异体HSCT。这些患者中观察到严重不良事件,包括CRS、多系细胞减少症和机会性感染。
CD7蛋白阻断剂。阻断T淋巴细胞中CD7表达的不同方法包括使用由单链可变片段和细胞内保留域组成的蛋白阻断剂,将目标抗原锚定在内质网和高尔基体中,然后进行蛋白降解。这种技术可以用来在不进行CD7基因编辑的情况下下调T细胞中的CD7表达。这种技术可以用来产生不在其细胞膜上表达CD7的功能和增殖的CAR-T细胞。Wong等人使用这种技术,最近报告了开发了既缺乏CD7又缺乏CD3表达的抗CD7 CAR-T细胞(PCART7),对T-ALL和T-淋巴瘤细胞表现出强大的细胞毒性。Zhang等人开发了一种基于串联CD7纳米体VHH6与内质网/高尔基保留基序的CD7阻断策略,以细胞内阻断CD7分子,从而促进其降解。临床前研究支持CD7阻断在预防同种异体杀伤和使用这种CD7阻断策略开发的CAR-T细胞对CD7阳性白血病细胞的强大细胞毒性方面的有效性。这些CAR-T细胞的I期临床试验显示,7/8的难治性/复发性T-ALL和T-淋巴瘤患者在CAR-T细胞输注后3个月实现了CR;1名患者实现了MRD阴性状态,一名T-淋巴瘤患者实现了超过12个月的CR。大多数患者只有1级或2级CRS。
Pan等人评估了供体来源的CD7 CAR-T细胞在治疗难治性/复发性T-ALL中的应用。为了最小化CD7 CAR-T细胞介导的同种异体杀伤,作者产生了一个包含抗CD7、4-1BB共刺激域和CD3ζ信号域的逆转录病毒载体,以及一个与内质网保留信号序列融合的CD7结合域,使CD7分子在细胞内保留。在I期临床试验中,从先前的HSCT供体或新供体生产的CD7 CAR-T细胞被用于20名难治性/复发性T-ALL患者。90%的患者实现了CR,其中七名患者在中位随访6.3个月后接受了HSCT,15名患者仍然处于缓解状态。2名患者(10%)出现了3-4级CRS,3名患者(15%)出现了1级神经毒性;这两种副作用都与T细胞供体类型或剂量水平无关。最近,这项试验的II期中期报告被提出,涉及20名难治性/复发性T-ALL患者的入组,中位随访11.0个月。90%的患者实现了CR;3名患者仍然处于缓解状态,7名患者复发,2名患者死于感染,8名患者接受了HSCT。1年的PFS和OS率分别为62%和60%。没有纵膈肿块的患者比有纵膈肿块的患者有更长的OS。在18名应答者中,有7名患者复发(3名CD7+,3名CD7-和1名未知)。
最近,Tan等人报告了CD7 CAR-T细胞I期研究中包含的T-ALL患者的长期(24-27个月)随访结果。在中位27个月的随访后,ORR和CRR分别为95%和85%,其中35%的患者接受了HSCT;20名患者中有6名复发,其中4名患者肿瘤细胞失去了CD7表达。在24个月的随访后,PFS和OS分别为37%和42%,中位PFS和OS分别为11和18.3个月。(表1)治疗后超过30天观察到的严重不良事件包括5次感染和1次4级肠道GvHD。在30天内,所有20名患者都出现了细胞减少症,而3名患者在输注后8、12.5和13个月出现了晚期3级细胞减少症。
在20名患者中有5人(25%)出现了非复发性死亡,主要原因是感染;在一例中,由于接受了SCT巩固治疗的患者发生了植入综合征。在所有患者中,非CAR CD7+ T和NK细胞在CD7 CAR-T细胞输注后15天内被清除,直到最后一次随访时在所有患者中几乎都无法检测到,除了一名患者。在2名患者中,长期监测T细胞表型显示中心记忆T细胞逐渐增加,并且在一名患者中,可以测量到低水平的原始和干细胞记忆T细胞亚群。
CAR-T细胞是通过自然CD7阴性T细胞或通过自然选择获得的CD7阴性CAR-T细胞生成的。从不表达CD7抗原的T淋巴细胞生成CAR-T细胞代表了一种潜在的替代CD7基因消除或阻断的方法。在这种情况下,Freiwa和同事提供了证据,表明自然发生的CD7- T细胞存在于健康受试者中,并且代表可以用于CAR-T细胞生成的功能效应T淋巴细胞。这些CD7- T细胞占T淋巴细胞的0.7-19%,主要具有CD4+记忆表型。从这些CD7-开始生成的CAR-T细胞主要表现出CD4+记忆表型,并且在体外和小鼠异种移植模型中对抗原暴露具有显著的抗肿瘤活性;重要的是,这些CAR-T细胞绕过了同种攻击。
Lu等人描述了通过体外自然选择过程获得CD7- CAR-T细胞的不同方法。特别是,这些作者比较了三种不同的方法来生成针对CD7的CAR-T细胞,并在临床前研究中评估了它们的特性:NS7CAR T细胞首先转导T细胞与CD7靶向载体,然后在两周的细胞培养过程中经历自然选择过程;Neg7CAR T细胞是通过转导CD3+CD7- T淋巴细胞与CD7靶向载体获得的;KO7CAR T细胞是通过CRISPR/CAS9基因编辑沉默CD7表达的T细胞生成的。与分选的CD7- CAR-T细胞和CD7敲除CAR-T细胞相比,NS7CAR T细胞显示出相似或更优的治疗特性,包括更高比例的CD8+记忆T细胞和更高比例的CAR+细胞。使用这些NS7CAR T细胞,对14名R/R T-ALL患者和6名T-LBL患者进行了I期临床研究;19名患者在骨髓中实现了MRD CR,5/9在髓外实现了CR。14名患者在接受NS7CAR-T细胞治疗后进行了allo-HSCT(10名巩固治疗,4名挽救治疗),没有复发;6名未接受allo-HSCT的患者中,有4名在中位时间54天时仍处于CR。只有一名患者经历了3级CRS。
Zhang和同事报告了在60名R/R T-ALL(35)和T-LBL(25)患者中使用CD7 CAR-T细胞NS7CAR在三个不同剂量水平:5x10^5/Kg、1-1.5x10^6/Kg和2x10^6/Kg治疗的结果。治疗后28天,94%的患者实现了骨髓CR;在32名EMD患者中,78%显示出客观反应,其中56%处于CR,22%处于PR;2年OS和PFS分别为63.5%和53.7%。重要的是,与未进行移植的10名患者相比,37名CR患者进行巩固HSCT的PFS明显更好(分别为67%和15%);在未进行移植的10名患者中,有8名复发。在有或没有EMD的患者中,OS和PFS没有观察到差异,而有之前移植史的患者显示出较低的1年OS率(49.4% vs 77.6%)。具有复杂细胞遗传学改变的患者与没有这些改变的患者相比,OS和PFS显著降低(分别为30% vs 79%和25% vs 64.2%);同样,携带TP53基因突变的患者显示出显著较低的OS(25% vs 77.9%)。安全性概况可接受,80%的患者出现1-2级CRS,11%的患者出现3-4级。两名患者(3.3%)经历了1级ICANS,1名患者(1.7%)经历了4级。CRS和ICANS的发生与NS7CAR的增殖无关。37名患者(61.7%)在输注后第30天或之后出现了未恢复的3级或更高级别的细胞减少。
自体和异体CD7 CAR-T细胞疗效的比较。Zhang等人对自体和异体CD7 CAR-T细胞治疗T细胞恶性肿瘤进行了比较分析。研究涉及10名R/RT-ALL和T-LBL患者,其中5名接受了自体CD7 CAR-T细胞治疗,5名接受了异体CD7 CAR-T细胞治疗;CAR-T细胞是Pan等人描述的Intrablock抗CD7 CAR-T细胞。尽管患者数量非常有限,但尝试了接受自体和异体CD7 CAR-T细胞治疗的患者之间的一些比较:CR率在异体中高于自体(分别为80%和40%);复发率在异体中高于自体患者(分别为100%和25%);CAR-T细胞在体内的存活率在异体中高于自体患者。
避免CD7 CAR-T细胞同种攻击的替代方法。最近,提出了两种避免CD7 CAR-T细胞同种攻击的替代方法,而无需进行基因组操作。Ye等人提出的一种方法是基于在制备CAR-T细胞期间,使用含有与CAR相同结合域的自由抗CD7单克隆抗体阻断T细胞膜上的CD7抗原。用抗CD7抗体培养的CAR-T细胞显示出抑制同种攻击、提高细胞活性,并能有效地介导对CD7阳性白血病细胞的细胞毒性。另一种方法是基于在这些细胞的体外扩增过程中向CD7 CAR-T细胞添加伊布替尼或达沙替尼,这些是关键CAR/CD3ζ信号激酶的药理学抑制剂:添加这些抑制剂可以挽救未编辑CD7 CAR-T细胞的体外扩增,在撤除抑制剂后恢复CAR-T细胞在体内介导的完全细胞毒性。使用这种方法制备的CAR-T细胞被证明适用于癌症治疗目的。
基于CD5靶向的T-ALL CAR-T细胞治疗。除了CD7,其他在正常和白血病T淋巴细胞上表达的膜抗原也是CAR-T细胞的合适靶标。其中之一是CD5;CD5在正常细胞上的表达仅限于T淋巴细胞和B1细胞。正如CD7的情况一样,CAR-T细胞上的CD5表达会导致CD5 CAR-T细胞的同种攻击。由于CD5靶向的CAR-T细胞治疗可能是治疗T细胞恶性肿瘤的一个有吸引力的策略,一些实验研究已经描述了针对CD5抗原的CAR-T细胞的特性。因此,Dai等人报告了开发具有完全人类重链-only抗原识别域的CD5靶向双表位CAR;通过CRISPR/CAS9基因编辑敲除CD5的T淋巴细胞与编码抗CD5的慢病毒载体转导生成的CAR-T细胞,在临床前模型中对白血病性淋巴T细胞展现出强大的细胞毒性。Ho等人提供了证据,表明CAR和CAR-T细胞上认知抗原的亲和力影响CAR-T同种攻击的强度。因此,研究表明,在CD5表达下调的T淋巴细胞中表达单链片段变量(scFv)会诱导低强度的同种攻击,从而产生具有最大化抗CD25效应活性的CAR-T细胞。另一项近期研究报告了开发和描述了通过CRISPR/CAS9基因编辑敲除CD5和CD7基因,以减轻CD5和CD7基因敲除的CAR-T细胞,表达抗CD5和抗CD7的完全人类重链-only域。这些CD5/CD7双特异性CAR-T细胞对T细胞恶性肿瘤展现出强大的抗肿瘤活性。很少有临床研究探索了CD5 CAR-T细胞在T细胞恶性肿瘤中的安全性和有效性。2019年,Hill等人报告了一项I期剂量递增研究(MAGENTA研究)的结果,该研究调查了在T细胞中表达时产生最小和短暂同种攻击的自体CD5导向CAR-T细胞。这种治疗被视为异体HSCT的桥梁。这项研究包括了9名患者,其中4名患者显示出客观反应,其中3名在血管免疫母细胞性T细胞淋巴瘤、外周T细胞淋巴瘤和T-ALL患者中实现了CR。输注后,PB CD3+细胞数量有所下降,但没有完全的T细胞无能。2021年,同一作者报告了在接受自体CD5 CAR-T细胞治疗的另外9名R/R T细胞淋巴瘤患者中观察到的结果。这四名反应患者中的三名随后进行了HSCT,其中两名分别在29个月和24个月后仍然存活并且处于CR。临床反应与输注的细胞剂量或T细胞扩增程度无关。Pan等人报告了一项I期研究的初步结果,该研究涉及治疗五名CD7阴性T-ALL患者在CD7 CAR-T细胞治疗后复发并接受了先前HSCT供体来源的CD5 CAR-T细胞;所有这5名患者在第30天实现了CR,并在中位随访27个月时保持MRD阴性。尽管这些结果是有希望的,但需要更长时间的随访来评估反应的持续时间和功能性免疫系统的重建。Patel和同事开发了一种新的CD5 CAR-T细胞制剂,Senza 5TM,这是一种自体CD5 CRISPR-CAS9敲除抗CD5 CAR-T产品,对CD5靶标具有高特异性;他们的策略,>90%的CD5 KO和>30%的CAR转导,允许生成两种主要的细胞群体:一种是CD5 KO CAR-T细胞,另一种是CD5KO正常未转导细胞。预计CD5 KO CAR-T5细胞将针对CD5阳性T-白血病/淋巴瘤细胞,但会导致对正常T细胞的毒性,这将通过在输注后具有生存优势的CD5 KO正常T细胞来减轻。将在I期临床研究中评估自体Senza 5TM CART5细胞在CD5阳性T细胞淋巴瘤患者中的效果。Chun等人最初报告了开发了一种通过CRISPR-CAS9基因编辑消除CD5表达的抗CD5 CAR-T细胞群体:CRISPR-CASp KO的CD5一致增强了CAR-T细胞的抗肿瘤活性,通过增加CAR介导的激活和增殖。
针对CD37的CAR-T细胞治疗T细胞恶性肿瘤。CD37是四跨膜蛋白超家族的一种跨膜蛋白。部分T细胞淋巴瘤在其细胞膜上表达CD37。Scarfs等人报告了以CD37为靶点的CAR-T细胞(CAR37)的开发,其中4-1BB作为共刺激域。CAR-T细胞在周围T细胞淋巴瘤模型中显示出抗原特异性激活、细胞因子产生和细胞毒性活性。在CAR-37细胞中未观察到显著的同种相残相关事件。迄今为止,唯一进行中的涉及CD37导向CAR-T细胞(NCT 04136275)的试验涉及CD37阳性血液系统恶性肿瘤的患者,包括白血病、B细胞和T细胞淋巴瘤。在初步的I期报告中,4名患者接受了CD37 CAR-T细胞治疗,其中包括1名CTCL患者,在输注后第28天实现了CR。
CD70靶向CAR-T细胞在T细胞淋巴瘤中的应用。CD70是一种II型跨膜糖蛋白,是Tnf配体家族的成员。CD70与其配体CD27相互作用。CD70表现出一些特性,使其成为某些血液系统恶性肿瘤的合适治疗靶点:CD70仅在激活的T和B细胞、NK细胞和树突状细胞上短暂表达;CD70在某些血液系统恶性肿瘤中广泛表达,包括一些B细胞淋巴瘤和系统性T细胞淋巴瘤;CD70与其配体CD27的相互作用在T和B淋巴细胞激活中诱导共刺激信号。
在患者衍生的异种移植模型中,使用抗体-药物偶联物靶向CD70在CTCL中产生了显著的抗肿瘤活性。
I期COBAL-LYM剂量递增研究评估了异体抗CD70 CAR-T细胞(CTX 130)在R/R患者中的PTCL或CTCL。CTX 130细胞是经过CRISPR/CAS9基因编辑改良的异体T淋巴细胞,以消除TCRα、MHC-I的表达,通过β2-微球蛋白破坏和CD70,然后转导含有编码抗CD70的慢病毒载体。15名T细胞淋巴瘤患者接受了CTX 130治疗;在PTCL(ORR 75%)和CTCL(ORR 67%)患者中观察到反应;29%的患者实现了CR。
T细胞受体靶向CAR-T细胞作为T淋巴瘤治疗策略。另一种针对T细胞恶性肿瘤的靶向策略基于T细胞受体β链恒定域1和2(TRBC1和TRBC2)的最终独占表达。正常T淋巴细胞包含TRBC1+和TRBC2+细胞组分,而T细胞恶性肿瘤仅限于其中之一。45靶向TRBC1的CAR-T细胞可以识别并杀死正常和恶性TRBC1+细胞,保留TRBC2+ T细胞。
最近,一项I/II期临床研究(AUTO4)的结果涉及使用TRBC1导向的自体CAR-T细胞治疗10名R/R T细胞淋巴瘤患者,采用四个平坦剂量水平。40%的患者实现了CR。最常见的治疗相关不良事件是细胞减少症(贫血和中性粒细胞减少症);然而,30%的患者有3级或更高的不良事件,PCR观察到没有CAR-T细胞扩增。随着随访时间的延长,接受最高剂量(450x10^6)治疗的50%患者在6个月和9个月时分别保持了完全的代谢反应。
使用CAR-T细胞治疗靶向CCR4在T细胞恶性肿瘤中的应用。趋化因子受体CCR4(也称为CD194)是一种七跨膜G蛋白偶联细胞膜分子,对造血系统细胞有选择性表达:特别是,CD4+CD25+Foxp3+调节细胞、TH2和TH7 T细胞主要表达CCR4。CCR4在许多T细胞恶性肿瘤中高度表达,包括ATL、CTCL、MF和SS,是这些疾病的生化治疗靶点。因此,mogamulizumab,一种去岩藻糖化的人性化抗体,经过工程改造以发挥增强的抗体依赖性细胞毒性,靶向CCR4,已被批准用于治疗R/R CTCL、MF、SS和ATLL。
这些观察结果为开发和评估靶向CCR4的CAR-T细胞提供了强有力的理由。因此,Perera等人产生了一个慢病毒载体,用于T细胞的遗传工程,以表达一个靶向CCR4的CAR,使用来自抗人CCR4抗体的人源化可变重链和κ轻链,与mogamulizumab不同。
CCR4靶向CAR-T细胞有效地溶解了患者衍生的CTCL细胞系,并在小鼠异种移植模型的ATL中发挥了强大的抗肿瘤活性。
Watanabe等人最近探索了靶向CCR4的CAR-T细胞的开发:在这些细胞的体外扩增过程中,同种相残事件特别耗尽了Th2和Tregs,同时保留了CD8+和Th1细胞。
在扩增过程结束时,产生了一种具有针对表达CCR4的T细胞恶性肿瘤的强大抗肿瘤效力的CAR-T细胞群。
CCR9在T-ALL中的靶向。最近的一项研究表明,CCR9在超过70%的T-ALL病例中表达,包括在R/R患者中超过85%的阳性率,与正常T细胞的低阳性率(<5%)相比。51靶向CCR9的CAR-T细胞对同种相残有抵抗力,并在体外和体内都具有强大的抗白血病活性,即使在抗原密度低的情况下。这些观察结果表明,抗CCR9 CAR-T细胞可能是R/R T-ALLs潜在的有希望的治疗策略。
靶向CD2的CAR-T细胞疗法。Xiang等人报告了一种异体“通用”CD2靶向CAR-T细胞(UCART2)的开发,其中CD2抗原被删除以防止同种相残,T细胞受体被移除以防止移植物抗宿主病(GvHD);UCART2细胞对T-ALL表现出显著的疗效,并延长了T-ALL植入的免疫缺陷小鼠的生存期。使用rhIL-7-hyFc(一种长效重组人IL-7),在原发性患者T-ALL模型中延长了UCART2的持续时间和增加了生存率。根据这些观察,建议异体抗同种相残的UCART2与rhIL7-hyFc联合使用,可能是治疗T-ALL的合适方法。
靶向CD38的CAR-T细胞疗法。最近的一项研究报告了靶向CD38抗原的CAR-T细胞的临床前活性。CD38抗原是多发性骨髓瘤和T-ALL中经过验证的肿瘤抗原。激活的T细胞上的CD38表达并未损害CD38-CAR-T细胞的扩增和体外功能。在用原发性T-ALL细胞异种移植的小鼠中,CD38-CAR-T细胞介导了延长的生存期。
结论
CAR-T细胞疗法治疗T细胞恶性肿瘤仍处于起步阶段,在其被引入这些疾病的标准治疗策略之前,还需要更多的研究。
有几个因素阻碍了CAR-T细胞技术在T细胞恶性肿瘤治疗中的成功开发:(i)肿瘤污染,与制造的CAR-T细胞产品与白血病/淋巴瘤T细胞的混合有关;(ii)T细胞再生障碍,作为不希望的靶向扩展到正常T细胞的后果;(iii)同种相残,与CAR-T细胞不仅靶向恶性T细胞,还靶向其他表达目标抗原的CAR-T细胞的细胞毒性有关。
已经开发了一些策略来消除或减轻这些限制;因此,异体CAR-T细胞绕过了CAR-T细胞与肿瘤细胞污染的问题。另一方面,已经开发了各种策略来减轻同种相残,从使用CRISPR-CAS9技术或基础编辑的基因编辑到细胞内阻断或自然选择。然而,这些策略中的一些解决了一个问题,但同时也引发了其他问题。
尽管存在这些限制,只有一些基于足够数量的T细胞恶性肿瘤患者、至少两年随访的研究,提供了初步的鼓励结果,这些结果需要在更大规模的前瞻性研究中得到确认。
未来的研究必须阐明:(i)CAR-T细胞在恶性T细胞表面要靶向的“最佳”膜抗原;(ii)自体和异体CAR-T细胞在安全性和有效性方面的比较评估;(iii)CAR-T细胞疗法单独或作为异体HSCT的桥梁的作用。